Abstract:
Recently, the team led by Academician Wei-Hong Zhu at East China University of Science and Technology proposed a novel photo-rearrangement reaction of diarylethenes induced by intramolecular proton transfer (IPT), while exploring the reactivity of active carbon sites in diarylethene systems. In this study, aldehyde and carboxyl groups were innovatively introduced at the active carbon positions, revealing distinct photoresponsive behaviors. The dialdehyde-substituted system exhibited typical reversible photochromism, whereas the dicarboxylate-substituted counterpart underwent an unexpected photorearrangement to yield polycyclic aromatic structures. Mechanistic studies, including transient absorption spectroscopy and density functional theory (DFT) calculations, demonstrated that the rearrangement proceeds through electrocyclization followed by an IPT-driven decarboxylation process. This work opens a new pathway for light-induced synthesis of polycyclic aromatic hydrocarbons and highlights potential applications in molecular modification, photoreaction mechanisms, and synthetic photochemistry.

Figure 1. Photo-rearrangement of diarylethenes induced by intramolecular proton transfer
Background:
Diarylethenes (DAEs), as a class of photoresponsive molecular switches, have attracted widespread interest in the field of photo-controlled functional materials. Structural modification of DAEs has mainly focused on terminal aryl positions, while directed functionalization at the inner carbon atoms—sites with dynamic reactivity during photoisomerization—has been less explored. Notably, during the photochemical isomerization from hexatriene to cyclohexadiene, the inner reactive carbon centers undergo significant changes in molecular orbital hybridization and bond angles. Thus, functionalizing these carbon centers represents a rational strategy for tuning the molecular design, photoresponsive behavior, and overall functionality of DAEs.
Highlights:
To address this, Academician Wei-Hong Zhu’s team investigated the modification of active carbon centers in two diarylethene systems, BBTE-ac and BBTE-al, which showed significantly different photochemical behaviors. Under light irradiation, the dialdehyde-substituted BBTE-al exhibited typical reversible photochromism. Surprisingly, BBTE-ac did not undergo the classic 6π-electron electrocyclization but instead underwent an unusual photorearrangement to afford a rearranged product, BBTE-ac2 (Figure 1). This product was monitored by NMR titration and fully characterized via single-crystal X-ray analysis of a protected derivative (BBTE-ac2boc, Figure 2).
Mechanistically, the photorearrangement begins with a typical electrocyclization to form a closed-ring intermediate featuring a six-membered hydrogen-bonded system. This is followed by an intramolecular proton transfer (IPT)-induced decarboxylation, yielding the final polycyclic aromatic product. The rearrangement was confirmed by femtosecond transient absorption spectroscopy (Figure 3) and further supported by DFT calculations (Figure 4). Interestingly, conversion of the carboxyl groups into amides restored the compound’s reversible photochromism, showcasing the versatility and tunability of the system for structural modification and synthetic photochemistry.
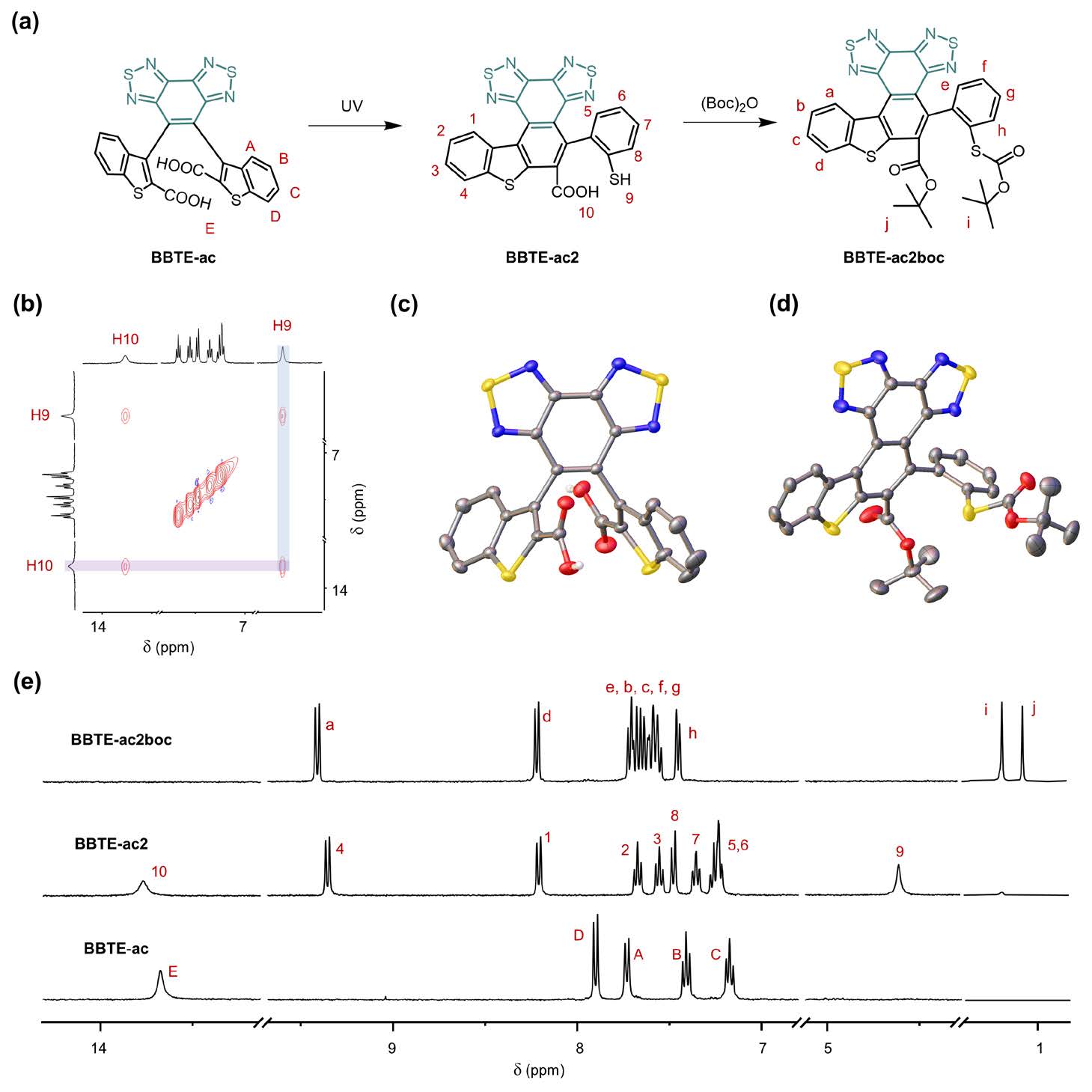
Figure 2. Characterization of the dicarboxylate-substituted BBTE-ac and its rearranged product
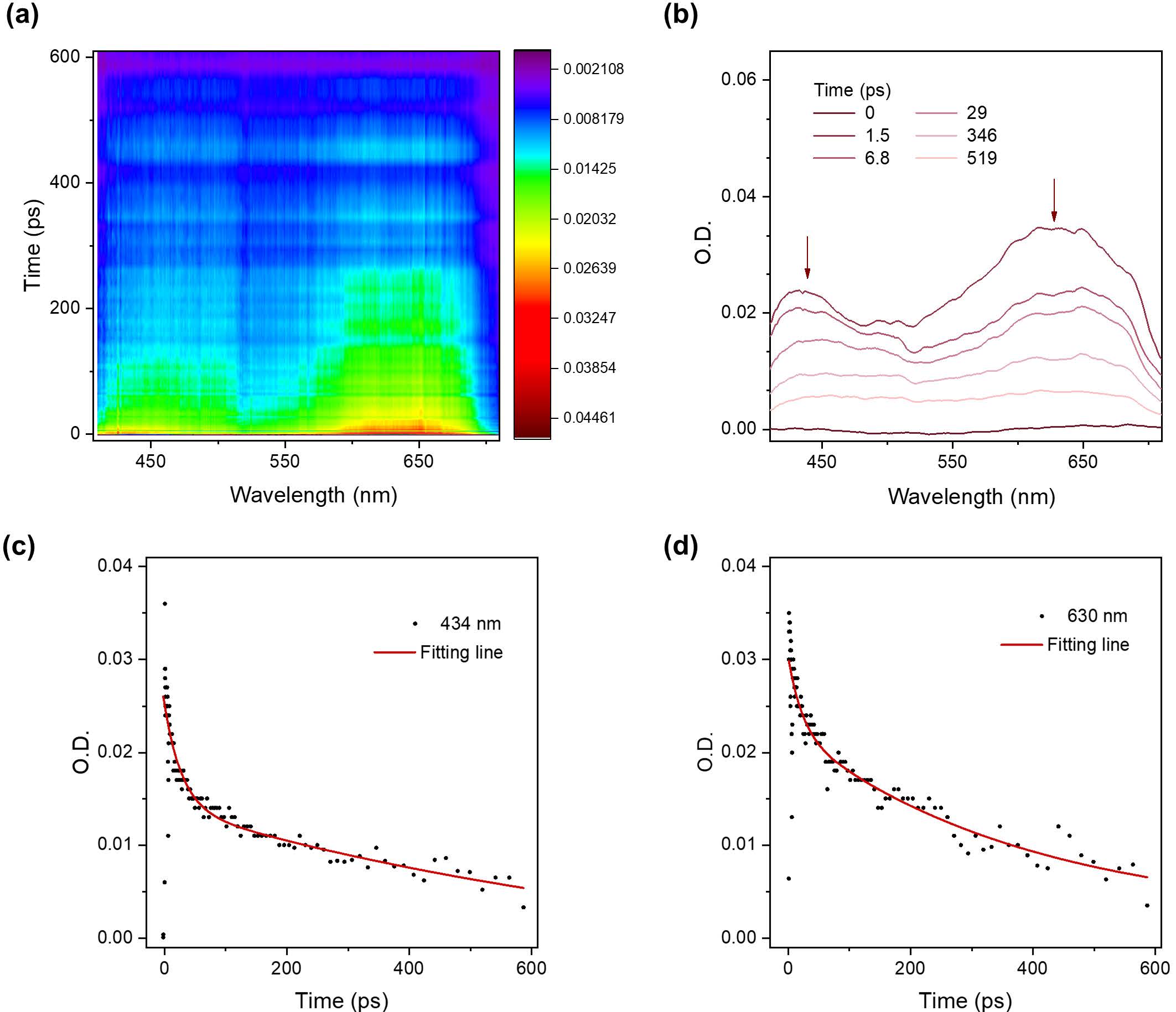
Figure 3. Ultrafast dynamics of BBTE-ac during photo-induced cyclization and rearrangement (on the picosecond timescale)
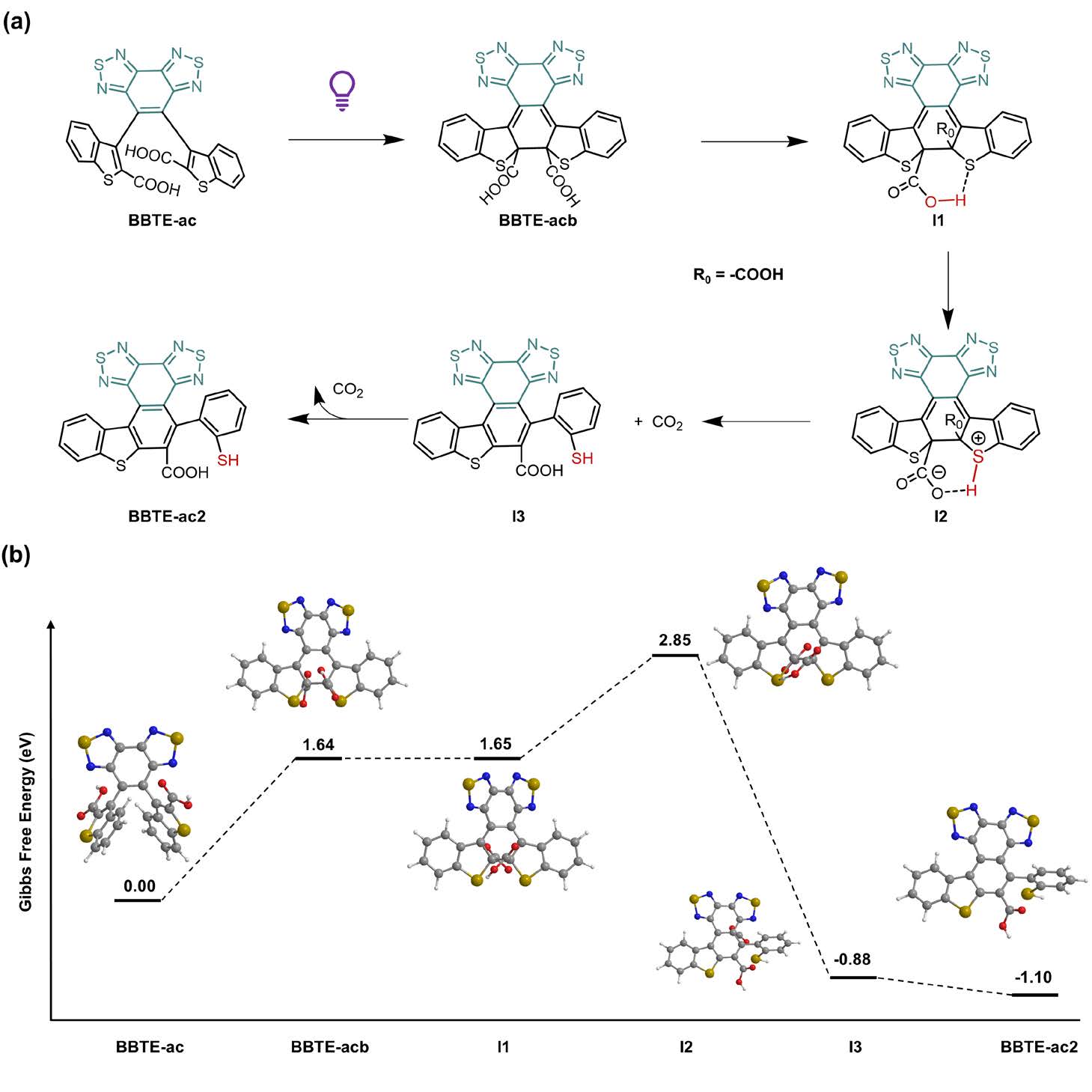
Figure 4. Mechanistic insights into the photo-induced rearrangement
Conclusion and Outlook:
In summary, this work reports the first observation of photo-rearrangement in diarylethenes via an intramolecular proton transfer mechanism and introduces a new synthetic strategy for polycyclic aromatic systems. By tuning the substituents at the active carbon sites, the authors achieved molecular engineering of DAE photoreactivity. While BBTE-al undergoes classic photocyclization under UV light, BBTE-ac displays a unique rearrangement yielding polycyclic aromatics. The mechanism involves a 6π-electrocyclization followed by IPT-induced decarboxylation, as revealed through multi-scale characterization and DFT analysis.
This discovery provides a novel concept for remote, non-contact photo-induced synthesis of polycyclic aromatic systems and holds significant promise for future applications in molecular design, photoreaction mechanism studies, and synthetic photochemistry.
This work was published as a Research Article in CCS Chemistry. Ning Lv is the first author. Academician Wei-Hong Zhu and Associate Professor Mengqi Li are co-corresponding authors. The research was supported by the National Natural Science Foundation of China through the Science Center Program, Major Research Plan Integration Project, and Key Projects.
Article Details:
Intramolecular Proton Transfer-Induced Photo-Rearrangement in Diarylethenes
Ning Lv, Zhiqiang Wang, Shaomeng Guo, Honglong Hu, Fanghui Li, Yifeng Chen, Mengqi Li*, and Wei-Hong Zhu*
Cite this by DOI: 10.31635/ccschem.025.202405382
Article Link: https://doi.org/10.31635/ccschem.025.202405382